Contents

A user’s guide
Alejandro R Jadad
2 Types of randomised controlled trials- There are many terms used to describe and classify RCTs.
- Some RCTs explore different aspects of the interventions.
- RCTs can be designed according to the participants' exposure to the interventions.
- RCTs can be classified according to the number of participants.
- RCTs can also be classified by the presence, absence, or degree of strategies to control bias.
- RCTs can take account of the preferences of eligible individuals.
Randomised clinical trials (RCTs) can be used to evaluate different types of interventions in different populations of participants, in different settings, and for different purposes. Once investigators ensure that allocation of participants to the study groups is random (to call the study an RCT), they can design the study using strategies to match the characteristics of the interventions they want to study, the resources they have available, and their academic, political, marketing, or clinical motivations.
Over the years, multiple terms have been used to describe different types of RCTs. This terminology has evolved to the point of becoming real jargon. This jargon is not easy to understand for those who are starting their careers as clinicians or researchers, because there is no single source with clear and simple definitions for all these terms.
In this chapter, I will describe the most frequent terms used to describe different types of RCTs. I will do my best to classify them in a way that will be easy for you to follow, understand, and remember. Some of the terms apply specifically to RCTs, whereas others may also be applied to other study designs. Some terms are mutually exclusive, some overlap considerably, and some complement each other. On occasion, I will include terms that are used to describe other types of studies that are not necessarily RCTs, to ensure that you are aware of the differences between them.
RCTs can be classified according to: (1) the aspect of the interventions investigators want to explore; (2) the way in which the participants are exposed to the interventions; (3) the number of participants included in the study; (4) whether the investigators and participants know which intervention is being assessed; and (5) whether the preferences of non-randomised individuals and participants are taken into account in the design of the study (Box 2.1).
RCTs that explore different aspects of the interventions
Depending on the aspects of the interventions that investigators want to evaluate, RCTs can be classified as: explanatory or pragmatic; as efficacy, effectiveness, or equivalence trials; and as phase I, II or III.
What is the difference between explanatory and pragmatic trials?
Explanatory trials address whether or not an intervention works. If the intervention works, then these trials attempt to establish how such intervention works. Typically, these trials are designed in such a way that the results are likely to yield a ‘clean’ evaluation of the interventions. To achieve this, the investigators set strict inclusion criteria that will produce highly homogeneous study groups. For instance, investigators designing an explanatory study of the effects of a new antihypertensive drug could decide to include only patients aged between 40 and 50 years, with no coexisting diseases (that is, diabetes) and exclude those receiving other particular interventions (b-blockers).
Explanatory trials also tend to use placebos as controls, fixed regimens (that is, 20 mg by mouth every 6 hours), long washout periods (if patients have been taking diuretics, for instance, those drugs will be stopped for a sufficient period of time to ensure that they are ‘washed out’ of their bodies), intention to treat analysis (see Chapter 3), and focus on ‘hard’ outcomes (that is, blood pressure recorded at specific times after a detailed and standardised process).
Pragmatic trials (also called management trials) are designed not only to determine whether the intervention works, but also to describe all the consequences of its use, good and bad, under circumstances mimicking clinical practice.1 To achieve this, pragmatic studies tend to use more lax criteria to include participants with heterogeneous characteristics, similar to those seen by clinicians in their daily practice. In addition to the more lax inclusion criteria, pragmatic trials tend to use active controls (that is, the new antihypertensive drug vs a b-blocker), flexible regimens (that is, 20 mg orally every 6 hours, reducing or increasing the dose by 5 mg according to the degree of blood pressure control and adverse effects), and analysis of the patients who received the interventions (see Chapter 3). Pragmatic trials do not preclude the use of ‘soft’ outcome measures, such as measures of sexual function or quality of life.
Although both explanatory and pragmatic approaches are reasonable and even complementary, it is important that you understand that they represent the extremes of a spectrum and that most RCTs include a combination of elements from each. The key issue is whether the investigators achieved the best combination of elements to answer their (and your) questions.
What is the difference between efficacy and effectiveness?
RCTs are often described in terms of whether they evaluate the efficacy or the effectiveness of an intervention. These two concepts are frequently misunderstood.
Efficacy refers to whether an intervention works in people who receive it.2 Trials designed to establish efficacy (also called efficacy trials) tend to be explanatory trials, because they are designed to yield a ‘clean’ evaluation of the effects of the intervention. In this particular case, however, the investigators are not so interested in finding out how the intervention works. Instead, their main goal is to include participants who will follow their instructions and who will receive the intervention. The extent to which study participants follow the instructions given by the investigators is called compliance or adherence. High compliance is easy to achieve when the administration of the interventions is completely controlled by the investigators or by other health professionals, who are not acting as investigators but are supportive of the study (that is, an RCT evaluating the effects of coronary artery bypass surgery and those of angioplasty in patients with unstable angina). This is more difficult when the interventions are not administered by the investigators but by the participants themselves, when the study has a long duration, and when the interventions have to be administered several times a day. Returning to the example of the antihypertensive drug discussed in the previous section, compliance will depend on the extent to which the participants take the antihypertensive tablets as prescribed for the whole duration of the study. The investigators in charge of this study may choose to include patients who have already shown high compliance in other studies.
Effectiveness refers to whether an intervention works in people to whom it has been offered.2 These RCTs, also called effectiveness trials, tend to be pragmatic, because they try to evaluate the effects of the intervention in circumstances similar to those found by clinicians in their daily practice. The design of effectiveness trials is usually simpler than the design of efficacy trials, because effectiveness trials tend to follow lax inclusion criteria, include flexible regimens, and allow participants to accept or reject the interventions offered to them. Typically, effectiveness trials evaluate interventions with proven efficacy when they are offered to a heterogeneous group of people under ordinary clinical circumstances.
On occasions, trials are designed not to detect possible differences in efficacy or effectiveness between two or more interventions, but to show that the interventions are, within certain narrow limits, ‘equally effective’3 or equally efficacious. These trials are called equivalence trials. Often, these trials seek to demonstrate that a new intervention (or a more conservative one) is at least as good as the conventional standard treatment. Investigators who engage in equivalence trials make efforts to minimise the risk of suggesting that the interventions have equivalent effects when in fact they do not. Strategies to minimise this type of risk are described in Chapters 3 and 4.
What are phase I, II and III trials?
These terms are used to describe the different types of trials that are conducted during the evaluation of a new drug. Phase I and II trials are not usually randomised.
As the name suggests, phase I trials are the first studies conducted in humans to evaluate a new drug. Phase I trials are conducted once the safety and potential efficacy of the new drug have been documented in animals. As the investigators know nothing about the effects of the new drug in humans, phase I trials tend to focus primarily on safety. They are used to establish how much of a new drug can be given to humans without causing serious adverse effects, and to study how the drug is metabolised by the human body.4 Phase I trials are mostly conducted on healthy volunteers. The typical participant in a phase I study is one of the investigators who developed the new drug, either an employee of a pharmaceutical company or a member of a research team at a university. People with diseases for which there is no known cure (that is, AIDS and certain types of cancer) often participate in phase I trials. As mentioned above, these trials are often not randomised, and not even controlled. Usually, they are just series of cases in which the participants are given incremental doses of the drug, without a control group, while they are monitored carefully by the investigators. In addition to the inherent limitations of case series, the main problem of this type of trial is that, if the participants are patients, those who are studied at the beginning are likely to receive very low doses which are unlikely to be effective, whereas those studied later are at greater risk of receiving toxic doses but are also more likely to benefit if the drug is effective.
After the safety of a new drug has been documented in phase I trials, investigators can proceed to conduct phase II trials. These are trials in which the new drug is given to small groups of patients with a given condition (usually about 20 per trial). The aim of phase II trials is to establish the efficacy of different doses and frequencies of administration. Even though phase II trials focus on efficacy, they can also provide additional information on the safety of the new drug. Often, phase II trials are not randomised, particularly when the therapeutic effects of the new drug can be measured objectively. For instance, if a new drug has been designed to treat a type of cancer that is associated with a high mortality rate, the investigators will conduct a phase II trial in which about 20 patients will receive the drug while tumour response, mortality and adverse effects are monitored carefully. If the drug is judged to be ineffective or excessively toxic, no more trials will be conducted. If the drug produces a good response (that is, ‘fewer patients than expected’ die), however, and patients tolerate its adverse effects, the investigators can proceed to a phase III trial. When the effects of the new drug are assessed using subjective measures (that is, pain relief with a new analgesic drug), the investigators can use a randomised design in which they will compare the effects of the new drug with a placebo (see below) to ensure that the effects observed in the small groups of patients can be attributed to the new drug and not to other factors (that is, a placebo effect).
Phase III trials are designed and conducted once a new drug has been shown to be reasonably effective and safe in phase II trials.4 Phase III trials are typically effectiveness trials, because they seek to compare the new drug with an existing drug or intervention known to be effective. This existing drug is usually regarded as the current standard treatment.4 Most phase III trials are RCTs.
There is an additional group of studies called phase IV trials. The term ‘phase IV trial’ is used to represent large studies3 that seek to monitor adverse effects of a new drug after it has been approved for marketing.4 These studies are also called postmarketing surveillance studies. They are mostly surveys and seldom include comparisons among interventions.3 The term ‘phase IV trial’ can also be used to describe promotional strategies to bring a new drug to the attention of a large number of clinicians.4 In either case, phase IV trials are not RCTs.
RCTs according to the participants' exposure to the interventions
Depending on the extent to which the participants are exposed to the study interventions, RCTs can have parallel, crossover, or factorial designs.
What is a parallel design?
Most RCTs have a parallel design. In these studies (also called parallel trials or RCTs with parallel group design), each group of participants is exposed to only one of the study interventions. For instance, if a group of investigators uses a parallel design to evaluate the effects of a new analgesic compared with those of a placebo in patients with migraine, they would give the new analgesic to one group of patients and placebo to a different group of patients.
What is a cross-over design?
An RCT has a cross-over design when each of the participants is given all the study interventions in successive periods. The order in which the participants receive each of the study interventions is determined at random. Crossover trials produce within participant comparisons, whereas parallel designs produce between participant comparisons. As each participant acts as his or her own control in crossover trials, they can produce statistically and clinically valid results with fewer participants than would be required with a parallel design.5
The time during which each of the interventions is administered and evaluated is called a period. The simplest crossover design includes only two periods. Returning to the example of the new analgesic, if the same group of investigators uses a crossover design, they would randomise each patient to receive the new analgesic first and then the placebo, or vice versa - the placebo first and then the new analgesic.
Crossover trials are not always appropriate. Every time you read the report of a crossover trial, you should explore the extent to which some basic rules are followed.
The interventions should be used in chronic, incurable diseases
Patients who are cured by one or more of the interventions will not be eligible to enter subsequent periods of a crossover trial. This means that the ability of the crossover trial to produce within patient comparisons is lost and the baseline characteristics of the participants in each period are no longer the same. Ignoring this rule can bias the results of a crossover trial substantially, particularly when a crossover trial is used to compare the effects of a drug that can cure a disease with those of a placebo6 (see Chapter 3).
The effects of interventions should have rapid onset and short duration
This minimises the risk of drop out within each period and helps to keep the number of participants stable across periods. In addition, if the effects of the intervention are of short duration, they are less likely to persist during the administration and evaluation of another intervention.
When the effects of an intervention are still present during the evaluation of another, such effects are called carry-over effects. If any observed difference between the interventions can be explained by the order in which the interventions were given to the participants, this is called a treatment-period interaction and it can invalidate the trial. Carry over effects can be predicted when the duration of the effects of the interventions are well known. In these cases, carry over effects can be prevented by separating the study periods by a period of time of sufficient length to enable the participants to be free of the influence of the intervention previously used by the time they receive the next intervention.3 This amount of time is also known as a washout period.
The condition (or disease) must be stable
If the disease is stable, the circumstances at the beginning of each period are more likely to be the same than if the disease is not stable. For instance, a crossover design to evaluate the new analgesic will produce more valid results in patients with migraine than in patients with postoperative pain because the intensity of postoperative pain tends to decrease with time. Even if the new analgesic was studied only in patients with migraine, the results of a crossover trial would be more valid if it included patients who have suffered similar episodes of migraine for many years than in patients who present episodes of migraine of different and unpredictable duration or intensity. All the differences between the study periods that are caused by disease progression, regression, or fluctuation are called period effects.
Carry over and period effects are known as order effects. Both can be assessed and removed from the comparisons by using statistical manoeuvres that are beyond the scope of this book, but that are described elsewhere.7
What is a factorial design?
An RCT has a factorial design when two or more experimental interventions are not only evaluated separately, but also in combination and against a control. For instance, a factorial design to study the effect of an opioid (that is, morphine) and a non-steroidal anti-inflammatory drug (that is, ibuprofen) for the treatment of cancer pain would mean that patients will be allocated randomly to receive ibuprofen only, morphine only, a combination of morphine and ibuprofen, or placebo. This design allows the investigators to compare the experimental interventions with the control (that is, morphine vs placebo), compare the experimental interventions with each other (that is, morphine vs ibuprofen), and investigate possible interactions between them (that is, comparison of the sum of the effects of morphine and ibuprofen given separately with the effects of the combination).
RCTs according to the number of participants
RCTs can include from one to tens of thousands of participants, they can have fixed or variable (sequential) numbers of participants, and they can involve one or many centres.
Is it possible for an RCT to have only one participant?
The answer is yes. These RCTs are called ‘n-of-1 trials’ or ‘individual patient trials’. Basically, they are crossover trials in which one participant receives the experimental and the control interventions, in pairs, on multiple occasions and in random order. These trials provide individual, rather than generalisable, results. They can be very useful when it is not clear whether a treatment will help a particular patient. You may find yourself in this situation, for instance, when you have a patient with a rare disease and there are no trials supporting the use of the treatment in that particular disease, or when the patient does not have a rare disease, but the treatment has been evaluated in studies that include very different patients.8 Typically, the number of pairs of interventions varies from two to seven. Usually, the number of pairs is not specified in advance, so that the clinician and the patient can decide to stop when they are convinced that there are (or that there are not) important differences between the interventions.
The success of n-of-1 trials to guide clinical decisions depends largely on whether the patient is willing to collaborate and on whether the rules described in relation to crossover trials (see above) are followed. A detailed description of how to design, conduct, and analyse n-of-1 trials is beyond the scope of this book, but can be found elsewhere.9
What is a mega-trial?
‘Mega-trial’ is a term that is being used increasingly to describe RCTs with a simple design (usually very pragmatic) which include thousands of patients and limited data collection.10,11 Usually, these trials require the participation of many investigators (sometimes hundreds of them) from multiple centres and from different countries. The main purpose of these large simple trials is to obtain ‘increased statistical power’ and to achieve wider generalisability. This means that their aim is to increase the chances of finding a difference between two or more interventions, if such a difference exists. This issue will be discussed in more detail in Chapter 5.
What is a sequential trial?
A sequential trial is a study with parallel design in which the number of participants is not specified by the investigators beforehand. Instead, the investigators continue recruiting participants until a clear benefit of one of the interventions is observed, or until they are convinced that there are no important differences between the interventions.12 These trials allow a more efficient use of resources than trials with fixed numbers of participants, but they depend on the principal outcome being measured relatively soon after trial entry.
What is a fixed size trial?
In a fixed size trial the investigators establish deductively the number of participants (also called sample size) that they will include. This number can be decided arbitrarily or can be calculated using statistical methods. The main goal of using statistical methods to calculate the sample size is to maximise the chance of detecting a statistically and clinically significant difference between the interventions when a difference really exists (see Chapter 5).
RCTs according to whether the investigators and participants know which intervention is being assessed
In addition to randomisation (which helps control selection bias), the investigators can incorporate other methodological strategies to reduce the risk of other biases. These biases and the strategies to control them will be discussed in detail in Chapter 3. I have brought this issue to your attention in this chapter because the presence, absence, or degree of one of these strategies has been used to classify RCTs. This strategy is known as ‘blinding’ or, perhaps more appropriately (but rarely used), ‘masking’. In clinical trial jargon, blinding or masking represents any attempt made by the investigators to keep one or more of the people involved in the trial (that is, the participant or the investigator) unaware of the intervention that is being given or evaluated. The purpose of blinding is to reduce the risk of ascertainment or observation bias. This bias is present when the assessment of the outcomes of an intervention is influenced systematically by knowledge of which intervention a participant is receiving. Blinding can be implemented at at least six different levels in an RCT. These levels include the participants, the investigators, or clinicians who administer the interventions, the investigators or clinicians who take care of the participants during the trial, the investigators who assess the outcomes of the interventions, the data analysts, and the investigators who write the results of the trial. As you might expect, in many studies those individuals who administer the interventions, take care of the participants, assess the outcomes, or write the reports are the same. Depending on the extent of blinding, RCTs can be classified as open, single-blind, double-blind, triple-blind, and quadruple-blind.
What is an open RCT?
An open RCT is a randomised trial in which everybody involved in the trial knows which intervention is given to each participant. Most trials comparing different surgical interventions or comparing surgery with medication are open RCTs.
What is a single-blind RCT?
A single-blind RCT is a randomised trial in which one group of individuals involved in the trial does not know the identity of the intervention that is given to each participant. Usually it is the participants or the investigators assessing the outcomes who do not know the identity of the interventions. Single blind designs are used frequently to evaluate educational or surgical interventions. For instance, investigators could evaluate different educational strategies in patients who would be unaware of the different types of strategies that are being compared. In addition, two surgical procedures could be compared under single-blind conditions with the use of identical wound dressings to keep investigators blind to the type of procedure during the assessment of the outcomes.
What is a double-blind RCT?
A double-blind RCT is a randomised trial in which two groups of individuals involved in the trial do not know the identity of the intervention that is given to each participant. Usually, these two groups include the participants and the investigators in charge of assessing the outcomes of the interventions.
To be successful, double-blinding requires that the interventions must be indistinguishable to both the participant and the investigator assessing the outcomes. Usually, the interventions are known to them just as A or B. When the experimental intervention is new and there are no standard effective interventions that could be used as controls, the investigators use an inert substance, or placebo, which has the same appearance and taste as the experimental intervention. These double-blind RCTs, in which the control group receives a placebo, are also called double-blind, randomised, placebo controlled trials.
When the RCT is designed to compare a new intervention with a standard treatment, the RCTs are called active-controlled. Achieving double-blinding in active-controlled trials is often difficult and frequently requires the use of what is called a double dummy. In a double-blind, double dummy RCT, each group of participants receives one of the active interventions and a placebo (in this case called a dummy) that looks and tastes the same as the other intervention. The double dummy technique is particularly useful when the investigators want to compare interventions that are administered by different routes or that require different techniques of administration. For instance, a double-blind, double dummy RCT would be the ideal study design to compare one intervention that is given as a tablet with another that is given by injection. In such a trial, the participants in one of the study groups would receive a tablet with the active drug and a placebo injection, whereas the participants in the other group would receive a placebo tablet and an injection with the active drug.
Problems with the way in which double-blinding is implemented, evaluated, and described in RCTs will be discussed in Chapters 3 and 4.
What is a triple-blind or quadruple-blind RCT?
In a triple-blind RCT, three groups of individuals involved in the trial do not know the identity of the intervention that is given to each participant. These groups could include the participants, the investigators giving the intervention, and those evaluating the outcomes (if the latter two are different); or the participants, the investigators evaluating the outcomes, and the data analysts.
If one more group is unaware of the identity of the intervention that is given to each participant, then the trial becomes a quadruple-blind RCT. This could easily be achieved by making the investigators who write the results of the trial unaware of the identity of the interventions until the time at which they complete the manuscript. Even though it is easy to make a double-blind trial into a triple or quadruple-blind trial, it happens very rarely.
RCTs that take into account the preferences of non-randomised individuals and participants
Eligible individuals may refuse to participate in trials, either because they have a strong preference for one particular intervention (if there are several active interventions available) or because they do not want to receive a placebo. Other eligible individuals may decide to participate in a trial despite having a clear preference for one of the study interventions. The outcomes of these individuals, whether they enter the trial or not, may be different from those participants who do not have strong preferences. The outcomes of the individuals who do not participate in the trials or of those who participate and have strong preferences are rarely recorded.
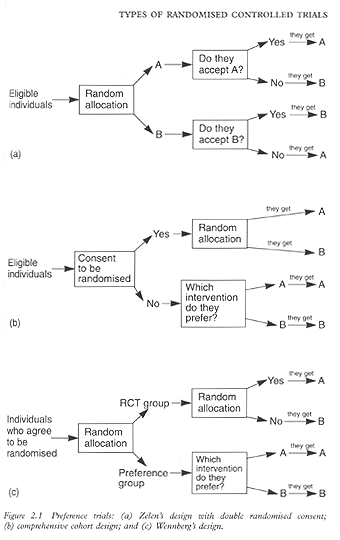
There are at least three types of RCTs that take into account the preferences of eligible individuals, whether they take part in the trial or not. These trials are called preference trials, because they include at least one group in which the participants are allowed to choose their own preferred treatment from among several options offered.13,14 These trials can have Zelen's design, a comprehensive cohort design, or Wennberg's design (Fig 2.1).
What is a trial with Zelen's design?
In a trial with Zelen's design, eligible individuals are randomised before they give consent to participate in the trial, to receive either a standard treatment or an experimental intervention. Those who are allocated to standard treatment are given the standard treatment and are not told that they are part of a trial, whereas those who are allocated to the experimental intervention are offered the experimental intervention and told that they are part of a trial. If they refuse to participate in the trial, they are given the standard intervention but are analysed as if they had received the experimental intervention.15
The main advantages of Zelen's design are that almost all eligible individuals are included in the trial and that the design allows the evaluation of the true effect of offering experimental interventions to patients. The main disadvantages are that they have to be open trials and that the statistical power of the study may be affected if a high proportion of participants choose to have the standard treatment.
To overcome the ethical concerns of not telling patients that they have been randomised to receive the standard treatment, the original approach proposed by Zelen can be modified by informing participants of the group to which they have been allocated and by offering them the opportunity to switch to the other group (Fig 2.1a). This design is also known as double randomised consent design.16 Even though this modified design overcomes the ethical concerns of the original Zelen's design, it does not solve the problems associated with lack of blinding and potential loss of statistical power.14
What is a trial with comprehensive cohort design?
A comprehensive cohort trial is a study in which all participants are followed up, regardless of their randomisation status (Fig 2.1b). In these trials, if a person agrees to take part in an RCT, he or she is randomised to one of the study interventions. If the person does not agree to be randomised because he or she has a strong preference for one of the interventions, that person will be given the preferred intervention and followed up as if he or she were part of a cohort study (see Chapter 7).16,17 At the end, the outcomes of people who participated in the RCT can be compared with those who participated in the cohort studies to assess their similarities and differences.
This type of design is ideal for trials in which a large proportion of eligible individuals are likely to refuse to be randomised because they (or their clinicians) have a strong preference for one of the study interventions.16 In these cases, it could be said that the study is really a prospective cohort study with a small proportion of participants taking part in an RCT.16 One of the main limitations of this type of design is that any differences in outcomes may be explained by differences in the baseline characteristics of the participants in the randomised and non-randomised groups.18,19
What is a trial with Wennberg's design?
In a trial with Wennberg's design eligible individuals are randomised to a ‘preference group’ or an ‘RCT group’ (Fig 2.1c). Those individuals in the preference group are given the opportunity to receive the intervention that they choose, whereas those in the RCT group are allocated randomly to receive any of the study interventions, regardless of their preference. At the end of the study, the outcomes associated with each of the interventions in each of the groups are compared and used to estimate the impact of the participants' preferences on the outcomes.
Preference trials are rarely used in health care research. They are, however, likely to become more frequently used as consumer participation in health care decisions and research increases.
References
1. Sackett DL, Gent M. Controversy in counting and attributing events in clinical trials. N Engl J Med 1979;301:1410-12.
2. Fletcher RH, Fletcher SW, Wagner EH. Clinical epidemiology: the essentials, 3rd edn. Baltimore, MD: Williams & Wilkins, 1996.
3. Armitage P, Berry G. Statistical methods in medical research, 3rd edn. Oxford: Blackwell Scientific, 1994.
4. Pocock SJ. Clinical trials: a practical approach. Chichester: Wiley, 1983.
5. Louis TA, Lavori PW, Bailar JC III, Polansky M. Crossover and self-controlled designs in clinical research. In: Bailar JC III, Mosteller F, eds. Medical uses of statistics, 2nd edn. Boston, MA: New England Medical Journal Publications, 1992:83-104.
6. Khan KS, Daya S, Collins JA, Walter SD. Empirical evidence of bias in infertility research: overestimation of treatment effect in crossover trials using pregnancy as the outcome measure. Fertil Steril 1996;65:939-45.
7. Senn S. Cross-over trials in clinical research. Chichester: John Wiley & Sons, 1993.
8. Guyatt G, Sackett D, Taylor DW, Chong J, Roberts RS, Pugsley S. Determining optimal therapyrandomized trials in individual patients. N Engl J Med 1986;314:889-92.
9.Sackett DL, Haynes RB, Guyatt GH, Tugwell P. Clinical epidemiology: a basic science for clinical medicine, 2nd edn. New York: Little, Brown & Company, 1991.
10. Woods KL. Mega-trials and management of acute myocardial infarction. Lancet 1995;346:611-14.
11. Charlton BG. Mega-trials: methodological issues and clinical implications. J R Coll Physicians Lond 1995;29:96-100.
12. Altman DG. Practical statistics for medical research. London: Chapman & Hall, 1991.
13. Till JE, Sutherland HJ, Meslin EM. Is there a role for preference assessments in research on quality of life in oncology? Quality of Life Res 1992;1:31-40.
14. Silverman WA, Altman DG. Patients' preferences and randomised trials. Lancet 1996;347:171-4.
15. Zelen M. A new design for randomized clinical trials. N Engl J Med 1979;300:1242-5.
16. Olschewski M, Scheurlen H. Comprehensive cohort study: An alternative to randomized consent design in a breast preservation trial. Methods Inform Med 1985;24:131-4.
17. Brewin CR, Bradley C. Patient preferences and randomised clinical trials. BMJ 1989;299:684-5.
18. Paradise JL, Bluestone CD, Rogers KD, Taylor FH, Colborn DK, Bachman RZ, Bernard BS, Schwarzbach RH. Efficacy of adenoidectomy for recurrent otitis media in children previously treated with tympanostomy-tube placement: Results of parallel randomized and nonrandomized trials. JAMA 1990;263:2066-73.
19. Torgerson DJ, Klaber-Moffett J, Russell IT. Patient preferences in randomised trials: threat or opportunity? J Health Services Res Policy 1996;1:194-7.
| Box 2.1 Different types of RCTs RCTs according to the aspects of the interventions they evaluate
|
|

Home | Contents | Foreword | Introduction | Acknowledgments | How to order
© BMJ Books 1998. BMJ Books is an imprint of the BMJ Publishing Group. First published in 1998 by BMJ Books, BMA House, Tavistock Square, London WC1H 9JR. A catalogue record for this book is available from the British Library. ISBN 0-7279-1208-9
![]()